- 移动端


上海杏园瑞民生物工程有限公司
11 年
手机商铺
- NaN
- 0
- 0
- 2
- 2
推荐产品





公司新闻/正文
Science | 微生物组单细胞测序新技术 Microbe-seq
1926 人阅读发布时间:2022-11-22 13:38

今年 6 月,哈佛大学和麻省理工学院的研究团队在 Science 上发表了题为 High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome 研究长文,介绍了他们在微生物群落研究方法学上取得的重大突破——Microbe-seq技术。
Microbe-seq 是一种具有菌株分辨率的高通量单细胞测序方法。Microbe-seq 技术集成了多种液滴微流控操作技术和定制开发的生物信息学分析手段,可应用于其他复杂的微生物群落,例如土壤和海洋中的微生物群落等。
微生物群落栖息于许多自然生态系统中,包括海洋、土壤和动物的消化道。微生物群落的行为和生物效应不仅取决于其组成,还取决于每个微生物群落内发生的生化过程以及它们之间的相互作用;这些过程强烈地受到生活在该群落中的每个微生物的基因组的影响。至今想要阐明微生物群落中的菌株水平信息,还面临相当大的挑战,如果能从微生物群落中解析出菌株水平的高质量基因组,那么这将极大地帮助我们理解微生物行为及其对宿主的影响。
目前,宏基因组测序技术被广泛应用到探索复杂微生物群落,但宏基因组学通常无法解析菌株水平的基因组。相反,基于培养的方法和基于滴度板的单细胞测序可以产生菌株解析的基因组,但只能获得有限数量的微生物菌株,且成本偏高。
Microbe-seq技术的发明解决了以上难题,它不需要培养即可从复杂微生物群落中获取成千上万个单细胞微生物的基因组信息,并组装出高质量的菌株水平基因组,从而能够在不损失分辨率或广泛物种适用性的基础上探究微生物群落的基因组。该方法应用面广泛,可用于具有复杂微生物群落的样本,如粪便、土壤和海洋等,在微生态研究中具有极大的市场应用潜力。
【技术原理】
实验原理:
1、利用液滴微流控技术,将成千上万的微生物单独地包裹在液滴中;
2、在每个液滴中,裂解微生物并释放 DNA;
3、进行全基因组扩增,将扩增出来的 DNA 打断并连上接头,随后在液滴中用带有特定序列的标签标记液滴内 DNA;
4、将所有液滴内的 DNA 合并,进行建库测序。
生信分析原理:
1、通过提取并比对各单细胞的基因组标志性信息,将样品中来自同一物种的单细胞微生物识别出来,然后合并在一起来组装出物种水平的参考基因组;
2、进一步将单个微生物的基因组和参考基因组对比,识别来自于不同菌株的单细胞微生物并进行基因组组装。
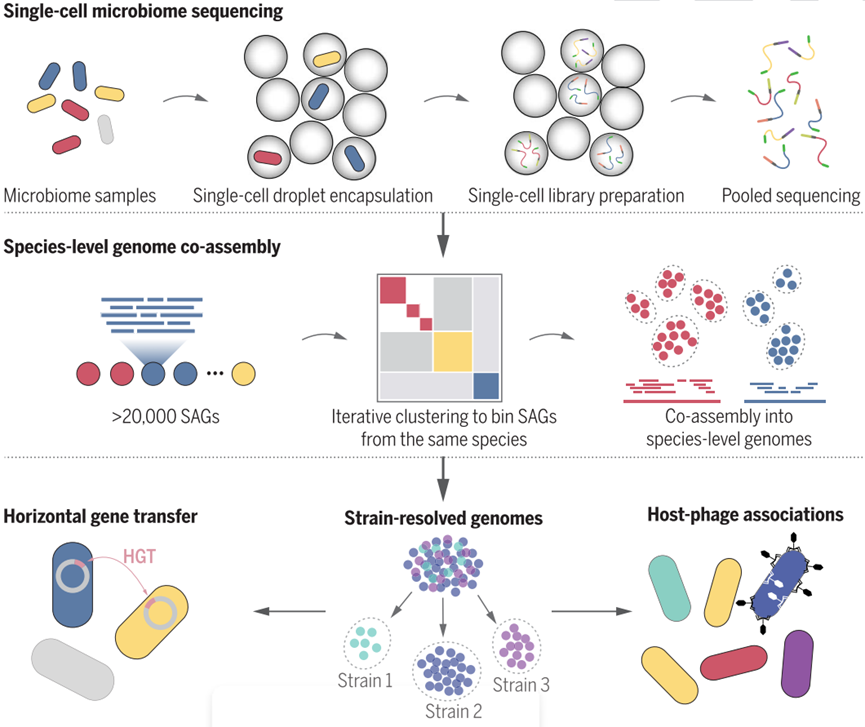
【技术优势】
- 高通量检测:一次可以获得成千上万单一微生物基因组,且无需培养
- 新物种、新菌株:发现新物种、新菌株,弥补宏基因组分辨不足的短板
- 基因组组装质量好:基因组质量与分离培养相同,支持功能基因/代谢通路研究
- 挖掘军逐渐关系:分析水平基因转移,研究宿主-噬菌体关联
【应用领域】
- 高精准度解析微生物群落结构
- 发掘有潜在应用价值的微生物资源
- 深入研究种间/种内的互相关系
- 深入研究病原菌的治病分子机制
- 单细胞水平解析耐药基因分布特征
- 改善复杂微生物组的参考基因组数据库
【Microbe-seq应用】
——基于前述Science论文
1.可在物种水平进行高质量基因组组装
- 来自一位健康人的7份粪便样本
- 组装了 76 个物种的中高质量基因组(见图1)
- 其中 52 个物种具有高等质量基因组
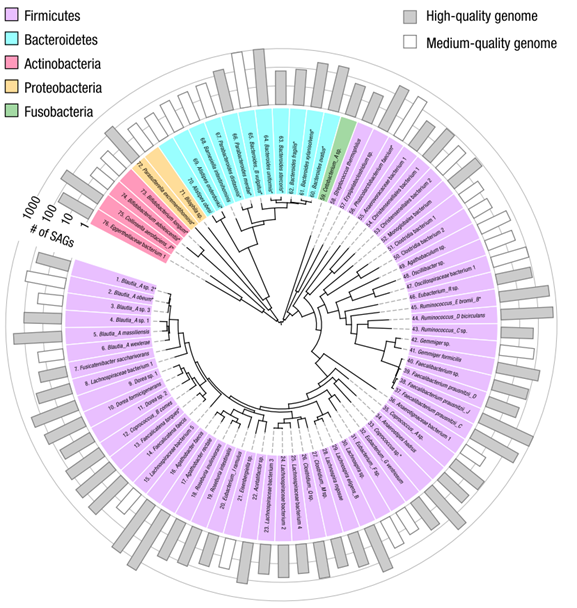
图1:单个人类供体的人类肠道微生物组中76种细菌的联合基因组。这 76 种细菌具有高质量或中等质量的共组装基因组。由核糖体蛋白序列构建的系统发育图由圆圈中心的树状图表示。每个物种的门由每个列出的物种名称后面的背景色表示(GTDB Tk数据库);从同一人类供体培养的分离物中获得基因组的19个物种用星号标记。用于共组装(丰度)的SAG数量由最外层环中的条表示,52个高质量基因组用灰色阴影表示,24个中等质量基因组用非阴影表示。
2.可获得媲美"金标准”水准的可靠基因组
- 其中 19 个物种已经被成功分离培养(见图1)
- 其中 17 个物种的共组装机因组与“金标准”基因组高度一致,ANI值超过99.5%以上
3.可获得宏基因组一致的微生物多样性
- 检测到的属同宏基因组结果重合度高达 96.9-99.8%
- 同一个门类的微生物的丰度趋势与宏基因组一致
- 通过共组装,获得 2 个菌株基因组(见图 2,菌株 A 和 C)和分离培养得到的基因组一致
- 还鉴定得到未能培养的全新菌株 B,并获得高质量基因组。
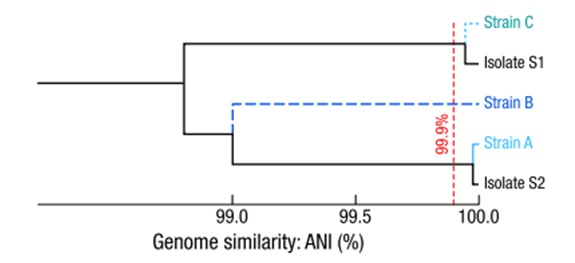
图 2:共同组装的普通双歧杆菌菌株高质量和中等质量基因组的系统发育,并与来自同一人类供体的分离株的相应基因组进行比较。树状图的水平轴表示这些菌株解析基因组之间的ANI值,表明共组装菌株 C 和分离株 S1 是同一菌株;同样,合集菌株A和分离菌株 S2 是相同的菌株。相比之下,第二丰富的菌株B在从同一人类供体培养的分离物中没有出现。
5.可在菌株水平分析水平基因转移事件
- 构建肠道微生物群中菌株的水平基因转移(HGT)网络(见图 3)
- 发现水平基因转移事件主要发生在同一细菌们的菌株之间
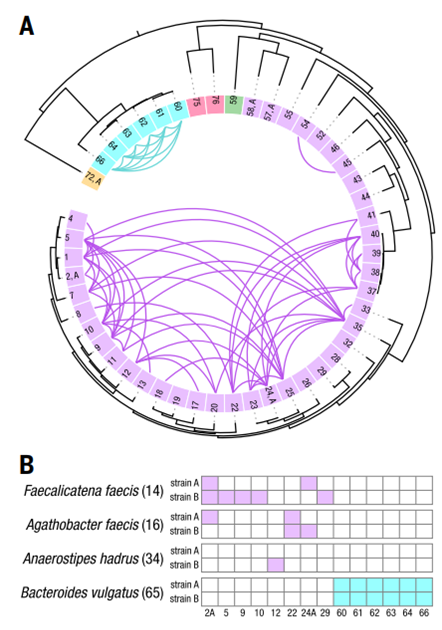
图3:单个供体的人体肠道微生物组内细菌菌株之间的 HGT。
(A) 在具有单个高质量菌株的49个物种中,HGT 解析了基因组,其顺序、数量和颜色如图2所示。检测到的两个基因组之间的HGT用一条曲线表示,其颜色与每个物种对的门的颜色相匹配。(B) 具有多个高质量菌株解析基因组的物种与具有单个高质量菌株分解基因组的物种之间的HGT,按照(A)中的编号。对于厚壁菌门中的细菌(屎肠杆菌、屎肠菌和哈德氏厌氧菌),每个菌株都具有不同种类的HGT。对于拟杆菌门,唯1的多菌种是普通双歧杆菌,它的两个菌株和该门中所有其他物种之间都有HGT。
(C) 共享HGT基因的物种数量分布。这些 HGT 序列中大约一半的基因在两个以上的物种中共享;几个基因存在于6或7种细菌菌株中。
6.可在菌株水平分析宿主-噬菌体关联信息
- 发现普通拟杆菌是噬菌体 crAssphage 的体内宿主物种(见图 4)
- 在菌株水平发现普通拟杆菌菌株A与噬菌体 crAssphage 存在显著关联
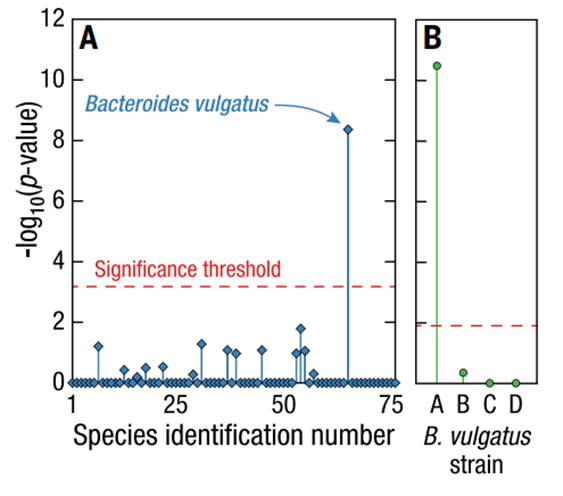
图4:人类肠道微生物组中宿主噬菌体与菌株特异性的关联。(A) 噬菌体 crAssphate 与具有高质量或中等质量基因组的细菌物种之间的关联,物种数量如图 1 所示。所有P值均通过单侧 Fisher 精确检验计算。与 crAssphate 显著相关的细菌物种是 B.vulgatus。(B) 四株普通芽孢杆菌与 crAssphate 的相关性。只有一种特殊的寻常型芽孢杆菌菌株——最丰富的菌株 A- 与 crAssphate 显著相关。